2023. 4. 21. 14:03ㆍ카테고리 없음
안녕하세요! 건강알리미입니다~

오늘은 ' 헌팅턴병[ Huntington's disease ] '에 대해 알아보고 정리하겠습니다!
헌팅턴병
[ Huntington's disease ]
-요약 -
보통염색체 우성으로 유전되며 무도증, 정신증상 및 치매가 주된 증상으로 나타나는 유전 질환
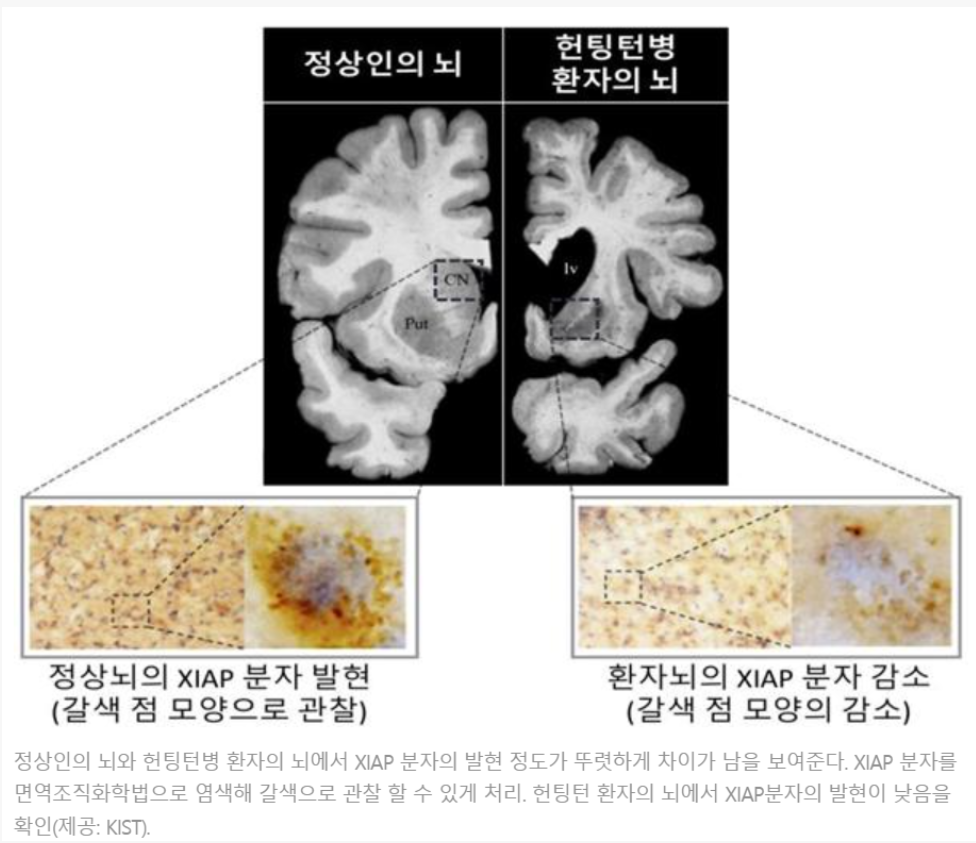
정의
염색체 4p16.3에 위치하는 헌팅턴(Huntington) 유전자에는 CAG 세 개의 염기가 반복되어 나타나는 특이한 서열이 존재한다. 헌팅턴병은 이 CAG 반복서열이 비정상적으로 증가되어 나타나는 질병으로 무도증, 정신증상 및 치매가 3대 증상이다. 병의 초기 단계에서는 무도증이 비교적 신체 일부에 국한되어 나타나지만 질환이 진행됨에 따라 무도증이 전신으로 퍼진다. 진단이 이루어진 후 사망에 이르기까지 15~20년 정도가 소요된다. 오랜 기간에 걸쳐 병이 진행되면 결국 과다근육긴장증과 심한 경직 상태에 이르게 되며 주로 흡인성 폐렴(음식물이나 입안의 미생물 등이 식도로 넘어가지 않고 기도로 잘못 흡인되어 발생하는 폐렴)으로 사망한다.

헌팅톤병
출처: 의학 · 간호 약어해설사전
원인
염색체 4p16.3에 위치하는 헌팅턴 유전자에는 CAG 세 개의 염기가 반복되어 나타나는 서열이 존재하는데, 헌팅턴병은 이 반복서열이 비정상적으로 증가되어 발생한다. 정상인에게서는 CAG 반복 횟수가 평균 19회 정도이지만 헌팅턴병 환자에게서는 40회 이상 나타나며, 반복 횟수는 헌팅턴병이 발병하는 나이와 반비례한다. 세대가 거듭될수록, 특히 부계 유전 시 대물림악화(anticipation)에 의하여 CAG 반복 횟수가 증가되면서 발병연령이 낮아지고 증상이 심해질 수 있다. 동일한 이상 증상이 산발성(명확히 밝혀지지 않은) 원인에 의해서도 발생할 수 있다.
증상
무도증, 정신증상 및 치매가 헌팅턴병의 3대 증상이다. 주로 30~40대에 발병하며 모계 유전인 경우 좀 더 늦게 발병하는 경향이 있다. 병의 초기 단계에서는 무도증이 비교적 신체의 일부에 국한되어 나타나지만 질환이 진행됨에 따라 전신으로 퍼진다. 오랜 기간에 걸쳐 병이 진행하면 결국에는 과다근육긴장증과 심한 경직(몸이 굳어서 뻣뻣해진 상태) 상태가 된다. 진단에서 사망까지는 15~20년이 걸리며 주로 흡인성 폐렴으로 사망한다. 정신 증상은 발병 전부터 나타날 수 있으며, 심한 우울증(30~50%)으로 인한 자살 가능성도 높다. 그 외에도 강박증, 자기조절력결핍 등의 증상이 나타날 수 있다.
진단/검사
신경과 의사의 진료를 통해 임상적 진단을 내린 후 유전자 검사 및 뇌 자기공명영상 촬영을 시행하여 진단할 수 있다.
헌팅턴병 환자의 뇌 영상검사 시 뇌 자기공명영상(뇌 MRI)상에서 미상핵(caudate nucleus)의 위축이 뚜렷하게 나타나고 기저핵(basal ganglia)의 대사 저하가 관찰된다.
치료
항불안제나 비정형 신경이완제 등의 약물을 투여하여 운동장애가 유발될 가능성을 최소화하면서 정신증상과 무도증을 완화시키도록 한다.
경과/합병증
병의 초기 단계에서는 무도증이 비교적 신체의 일부에 국한되어 나타나지만 병이 진행되면서 전신으로 퍼진다. 오랜 기간에 걸쳐 병이 진행되면 결국에는 과다근육긴장증과 심한 경직(몸이 굳어서 뻣뻣해짐) 상태가 나타나게 된다. 진단에서 사망까지는 15~20년이 소요되며 주로 흡인성 폐렴으로 사망한다.
예방방법
특별한 예방방법은 없다.
문제
A 40-year-old man is brought to the emergency department after sustaining multiple lacerations during a bar fight. The patient’s wife says that he has been showing increasing aggression and has been involved in many arguments and fights over the past 2 years. He has no significant past medical or psychiatric history and currently takes no medications. He cannot provide any relevant family history since he was adopted as an infant. Vital signs are within normal limits. On physical examination, the patient looks apathetic and grimaces repeatedly. Suddenly, his arms start to swing by his side in an uncontrolled manner. Which area of the brain is most likely affected by this patient’s disease process?
|
Result:
Explanation:
Correct answer A: This patient is showing signs and symptoms of Huntington disease (HD), an autosomal dominant, progressive, fatal, neurodegenerative disorder. A family history of HD is almost always present as sporadic cases are extremely rare. Symptoms typically onset in the 30s or 40s. The disease is rapidly progressive and is generally fatal within 20 years of diagnosis.
Typical clinical signs and symptoms include:
|
Movement/muscular impairments
|
|
|
Cognitive problems
|
|
|
Psychiatric complications
|
|
The underlying genetic cause of the disease is the expansion of CAG trinucleotide repeats within the Huntingtin gene on chromosome 4. This mutation leads to an irreversible loss of GABAergic neurons in the striatum (caudate nucleus and putamen), leading to an imbalance in GABA and dopamine levels in the basal ganglia.

Image: The basal nuclei - the most common brain structure damaged in Huntington disease. By Lecturio
Option B: Cerebellar disease leads to various motor problems like ataxia, loss of coordinated movement, dysarthria, dysdiadochokinesia, dysmetria, hypotonia, nystagmus, and intention tremor. This patient’s presentation is more consistent with HD than a cerebellar pathology.
Option C: Damage to the cortical areas responsible for movement usually leads to upper motor neuron signs and symptoms (rigidity, increased muscle tone, spasticity).
Option D: Lesions in the medulla oblongata lead to the loss of function of cranial nerves IX-XII as these originate from the medulla. This area is not affected by Huntington disease.
Option E: Parkinson disease is associated with damage to dopaminergic neurons in the substantia nigra pars compact. It presents in older adults with tremor, bradykinesia, and shuffling gait.
Learning objective: Huntington disease (HD) is an autosomal dominant rapidly progressive neurodegenerative disorder. A family history of HD is almost always present. Typical clinical signs and symptoms include motor, cognitive, and psychiatric impairments.
|
Related Videos:
|
|
Book References:
First Aid for the USMLE Step 1 (2022, 32nd ed): 538 First Aid for the USMLE Step 1 (2021, 31st ed): 537, 538 First Aid for the USMLE Step 1 (2020, 30th ed): 520 First Aid for the USMLE Step 1 (2019, 29th ed): 508 First Aid for the USMLE Step 1 (2018, 28th ed): 504 First Aid for the USMLE Step 1 (2017, 27th ed): 490 |
오늘은 여기까지 정리하겠습니다!
감사합니다!
